- How to code a functional molecular machine?
-
Date
2018-05-30 -
- How life evolves the genes that code for molecular machines like protein -
The strategy of their study, which was published in Proceedings of the National Academy of Sciences (PNAS), is to construct a simple conceptual framework to examine and understand the genetic coding of protein and how evolution searches and finds these codes effectively. They built a simple model of protein based as an elastic material. As shown in Figure 1, the protein is made of red and blue amino acids connected by molecular “springs”. The red amino acids are flexible while the blue ones are rigid. Therefore, higher occurrence of the red amino acids at the center of the protein gives rise to a floppy channel within the protein, which allows the protein to perform large scale “hinge” movements as shown in the Figure. This motion allows them to bind effectively to other molecules.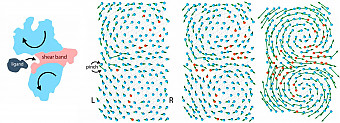
Figure 1: (A) When a protein binds to a ligand it undergoes large scale motion due to the presence of certain “floppy” region (red “shear band” ) across the protein. This “floppy” region separated the rigid blue regions of the protein. These large movements are signatures of functional proteins. (B)-(D) shows different stages of evolution of a protein: from a non-functional (B) to a functional protein (D). The protein is modeled as an elastic spring network with two kinds of amino acids: red amino acids are flexible and blue amino acids are rigid. Binding to a ligand “pinches” the protein at the binding site. However since the protein is mostly rigid (blue) the protein can not move making it non-functional. (C) During evolution more flexible (red) amino acids are added and it functions better. (D) The “floppy” (red) region forms at the center of the protein. The protein can move and bind to ligands easily.
During evolution, the 0’s and 1’s in the gene are randomly flipped through a process called mutation. Most mutations end in non-functional proteins, however some rare mutations can give rise to a functional protein as in Fig 1D. How does evolution “know” how to form functional proteins? Essentially both functional and non-functional proteins are produced during evolution, however thanks to “the survival of the fittest” only the functional proteins are kept and the non-functional proteins eventually die out. In the current model, the motion or the displacements of the protein during the binding measures this fitness, how well they perform their biological function of binding. The fittest functional proteins show large movements. Using simple simulations with this “motion-fitness” of the principal concept of the survival of the fittest, the authors have found what kind of mutations will end in functional proteins.
The “floppy” channel in the protein has another interesting and peculiar consequence. Any flipping of 0’s or 1’s at one end of the channel strongly affects the flipping at the other end of the channel. This correlation effect, called epistasis, is thus very long ranged and can affect amino acids along the channel and might provide insight into the drug development in medicine.
In the future, the research team aims to explore some possible application of their findings to certain real proteins like kinases. The study open avenues to investigate the evolution of other functions of proteins like molecular recognition. Huge databases, which have been developed through years of research already exist on the evolution of proteins. Some simple underlying phenomena can probably be uncovered using the current theory.Journal Reference: Sandipan Dutta, Jean-Pierre Eckmann, Albert Libchaber, and Tsvi Tlusty. Green function of correlated genes in a minimal mechanical model of protein evolution. PNAS (2018). DOI: 10.1073/pnas.1716215115
